
背景及び目的
酵素と阻害剤のドッキング計算を行う際には、従来の古典的手法では記述できない、電子的な相互作用が重要となる場合があります。量子化学計算手法であるフラグメント分子軌道(FMO)法では、古典的パラメタに依存することなく電子の挙動を定量的に扱うことができます。
解析事例
現在、肺がんの分子標的薬であるEGFRチロシンキナーゼ阻害剤には、イレッサなどが用いられていますが、薬剤耐性などが課題となっています。そこで、水素結合よりも結合の強い不可逆的阻害剤の設計として、ホウ素化合物に着目したドッキング計算および結合エネルギー解析を行いました。
EGFRとTarcevaの複合体構造
成果及び展望
阻害剤であるTarcevaと、候補化合物のドッキング構造(Model1~Model3)について結合エネルギーの計算を行った結果、ホウ素の空軌道のsp3結合性を用いたModel1の結合が最も強いという結果が得られました。このように量子化学計算手法を用いることで、古典的パラメタの決定が難しい化合物に関しても、計算による結合性の予測や相互作用の解析を行うことができます。
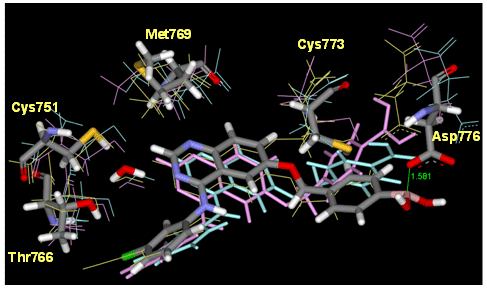
阻害剤の結合様式
左右スクロールで表全体を閲覧できます
| Tarceva | Mode1 | Mode2 | Mode3 | |
|---|---|---|---|---|
|
HF |
-33.6 |
-90.2 |
-50.9 |
-56.3 |
|
MP2 |
-100.3 |
-154.0 |
-105.9 |
-116.6 |
結合エネルギー
参考文献
H. S. Ban, T. Usui, W. Nabeyama, H. Morita, K. Fukuzawa, and H. Nakamura “Discovery of Boron-conjugated 4-Anilino-quinazoline as a Prolonged Inhibitor of EGFR Tyrosine Kinase” Org. & Biomol. Chem.. 7, 4415-4427 (2009).
お問い合わせ
担当:サイエンスソリューション部