
Quality control(QC)解析
csDAIのQC解析ではスプレッドシート等で開いて確認することが出来るタブ区切りのテキストファイルと様々な項目の図、日本人で頻度の高いSNPsによる遺伝子型一致割合が出力されます。
Quality control解析で出力されるファイル
左右スクロールで表全体を閲覧できます
| ファイル名 | 内容 |
|---|---|
|
ラン名.QC.pdf |
サンプル毎及びlane毎に表示した3種類の表とサンプル毎及びlane毎1ページに16個の図が作られる。Lane毎のQCの結果をマージした場合はマージ後の統計量の表とサンプル毎に4つの図が作られる。 |
|
ラン名.QC.tsv |
各種統計量がサンプル及びlane毎に出力される。Laneのマージがあった場合はマージ後の統計量がタブ区切りで出力される。 |
|
ラン名.concordance.tsv |
同じラン内の全てのサンプルペアについて日本人で比較的頻度の高いSNPsにおける遺伝子型一致割合が出力される。サンプル数が1の場合は出力しない。 |
|
ラン名.concordance.pdf |
上記遺伝子型一致割合のヒストグラム。ただしサンプル数が3未満の場合は出力しない。 |
QCの表(テキストファイル)
テキストファイルに出力されるQC項目
左右スクロールで表全体を閲覧できます
| 項目 | 内容 |
|---|---|
|
SampleID |
サンプルID |
|
experimetName |
ラン名 |
|
lane |
レーン番号 |
|
#cluster |
クラスター数 |
|
#PF |
パスフィルター通過数 |
|
%PF |
パスフィルター通過数×100/クラスター数 |
|
#identical |
A,T,G,C,N完全一致paired-end read数 |
|
%identical |
A,T,G,C,N完全一致paired-end read数×100/パスフィルター通過数 |
|
#paired-end |
A,T,G,C,N完全一致paired-end read削除後のpaired-end read数 |
|
#adaptored |
skewer*1によりアダプター配列が除かれたpaired-end read数 |
|
%adaptored |
skewer*1によりアダプター配列が除かれたpaired-end read数×100/#paired-end |
|
#removed |
skewer*1によりアダプター配列が除かれた後に25bp未満となり削除されたpaired-end read数 |
|
#remain-paired-end |
skewer*1によるアダプター配列削除後のpaired-end read数 |
|
read1-N% |
Read-1のNの割合(%) |
|
read1-A% |
Read-1のAの割合(%) |
|
read1-T% |
Read-1のTの割合(%) |
|
read1-G% |
Read-1のGの割合(%) |
|
read1-C% |
Read-1のCの割合(%) |
|
read2-N% |
Read-2のNの割合(%) |
|
read2-A% |
Read-2のAの割合(%) |
|
read2-T% |
Read-2のTの割合(%) |
|
read2-G% |
Read-2のGの割合(%) |
|
read2-C% |
Read-2のCの割合(%) |
|
read1-meanQ |
Read-1の全平均Q-value |
|
read1-QA |
Read-1のAの平均Q-value |
|
read1-QT |
Read-1のTの平均Q-value |
|
read1-QG |
Read-1のGの平均Q-value |
|
read1-QC |
Read-1のCの平均Q-value |
|
read2-meanQ |
Read-2の全平均Q-value |
|
read2-QA |
Read-2のAの平均Q-value |
|
read2-QT |
Read-2のTの平均Q-value |
|
read2-QG |
Read-2のGの平均Q-value |
|
read2-QC |
Read-2のCの平均Q-value |
|
%NotClipped |
bwa mem*2もしくはSTAR*3でsoft clip及びhard clip無しでマップされたpaired-end readの割合(%) |
|
%Clipped |
bwa mem*2もしくはSTAR*3でsoft clip若しくはhard clip有りでマップされたpaired-end readの割合(%) |
|
%NotClippedEdit |
bwa mem*2もしくはSTAR*3でsoft clip及びhard clip無しでマップされたpaired-end readのedit distanceの平均値 |
|
%NotClippedMiss |
bwa mem*2もしくはSTAR*3でsoft clip及びhard clip無しでマップされたpaired-end readのミスマッチ割合の平均値 |
|
%ClippedEdit |
bwa mem*2もしくはSTAR*3でsoft clip若しくはhard clip有りでマップされたpaired-end readのedit distanceの平均値 |
|
%ClippedMiss |
bwa mem*2もしくはSTAR*3でsoft clip若しくはhard clip有りでマップされたpaired-end readのミスマッチ割合の平均値 |
|
removeTileThreshold |
不良タイルとして削除する時の、NotClippedマッピング割合の閾値 |
|
removedTiles |
不良タイルとして削除されたタイルとそのタイルのNotClippedマッピング割合 |
|
InsertMean |
平均insert長(BAMのマージではマッピングされた全read、それ以外ではsoft clip無しでマッピングされたreadのみ) |
|
InsertSD |
Insert長の標準偏差(BAMのマージではマッピングされた全read、それ以外ではsoft clip無しでマッピングされたreadのみ) |
|
InsertMedian |
Insert長の中央値(BAMのマージではマッピングされた全read、それ以外ではsoft clip無しでマッピングされたreadのみ) |
|
#Mapped |
samtoolsでカウントされた総single-end read数 |
|
#PCR-duplicates |
samtoolsでカウントされたPCR duplicatesフラグがあるsingle read数 |
|
%PCR-duplicates |
#PCR-duplicates×100/#Mapped |
|
#Properly-paired |
samtoolsでカウントされたproperly pairedフラグがあるsingle read数(PCR duplicateフラグ有りは除く) |
|
%Properly-paired |
#Properly-paired×100/#Mapped |
|
#chrX |
samtoolsでカウントされたX染色体にマップされた総single read数(PCR duplicateフラグ有りは除く) |
|
#chrY |
samtoolsでカウントされたY染色体にマップされた総single read数(PCR duplicateフラグ有りは除く) |
|
%chrX |
#chrX×100/#Mapped |
|
%chrY |
#chrY×100/#Mapped |
|
Contamination |
verifyBamID*4による他サンプルコンタミネーション割合の推定 |
|
bait(DNA-seqのみ) |
Baitファイル名 |
|
#On-bait |
samtools*5でカウントされたbait領域内にあるproperly pairedのsingle read数(PCR duplicateフラグ有りは除く) |
|
%On-bait |
#On-bait×100/#Properly-paired |
|
meanDepth |
Properly pairedのみによるbait領域内の平均depth(PCR duplicatesは除く) |
|
medianDepth |
Properly pairedのみによるbait領域内のメディアン値(PCR duplicatesは除く) |
|
80-thDepth |
Properly pairedのみによるbait領域内の80% percentile値(PCR duplicatesは除く) |
|
unstranded |
STAR*3のReadsPerGene.out.tabに出力されるENSGにマップされたread数(GTF中に両ストランドにexonがある領域はambiguousにカウントされるため除かれる)。 |
|
sense |
STAR*3のReadsPerGene.out.tabに出力されるENSGにマップされた1st read数 |
|
anti-sense |
STAR*3のReadsPerGene.out.tabに出力されるENSGにマップされた2nd read数 |
|
MALAT1ratio5 |
MALAT1(noncoding RNA)の5'側のreadカウント数/3'側のreadカウント数 |
|
MALAT1ratioM |
MALAT1(noncoding RNA)の中点のreadカウント数/3'側のreadカウント数 |
|
KMT2Dratio5 |
KMT2D遺伝子の5'側のreadカウント数/3'側のreadカウント数 |
|
KMT2DratioM |
KMT2D遺伝子の中点のreadカウント数/3'側のreadカウント数 |
|
heteroReadFreq |
日本人で頻度0.5程度のSNPsの内、heteroとcallされた箇所の平均alternative read数の割合 |
|
heteroReadSD |
日本人で頻度0.5程度のSNPsの内、heteroとcallされた箇所の平均alternative read数のSD |
|
heteroReadCV |
heteroReadSD/ heteroReadFreq |
|
versions |
QC解析で使用したソフトウエアのバージョン情報 |
- *1)
- *2)
- *3)
- *4)
- *5)
QCレポートの図
PDFファイルに出力される図の説明
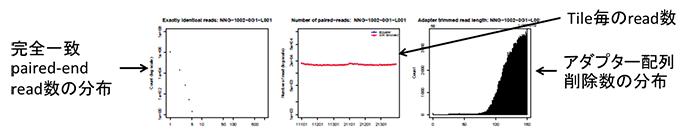
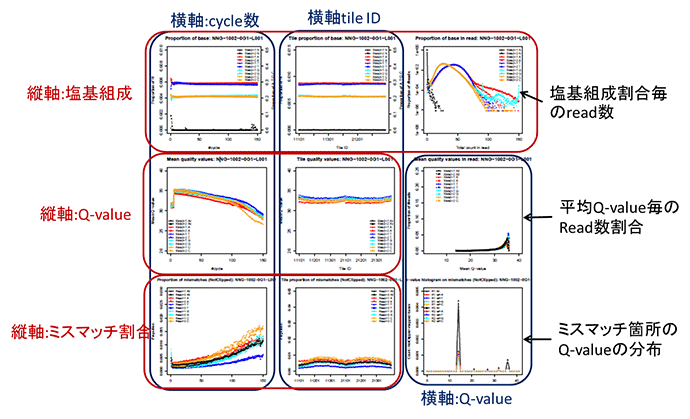
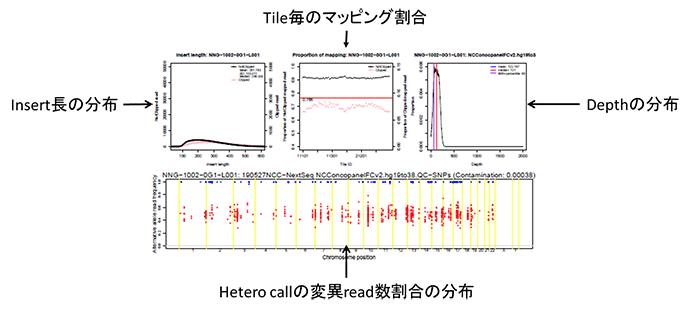
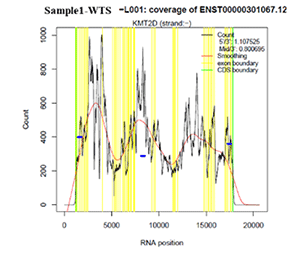
RNA-seqの場合はbait領域におけるdepthのヒストグラムの代わりにENST00000301067.12(KMT2D)遺伝子上でのdepthの分布が挿入される。
末梢血サンプルとFFPE(formalin-fixed paraffin-embedded)検体の違いによる典型的な図の変化を下記に示しました。
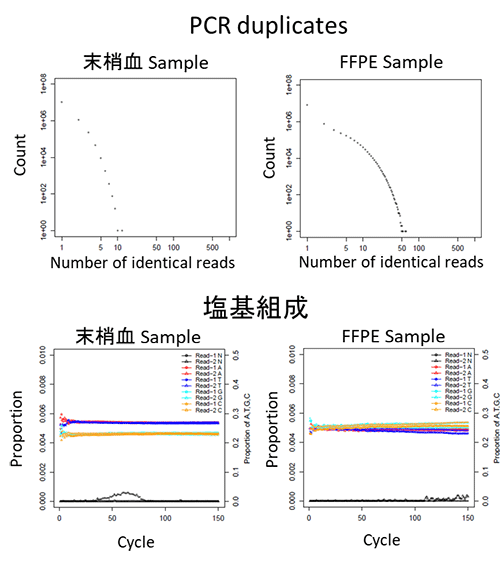
Read長が長すぎる場合、特にread-2で塩基callの品質が落ちることがあります。
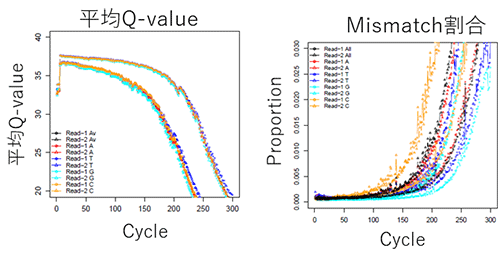
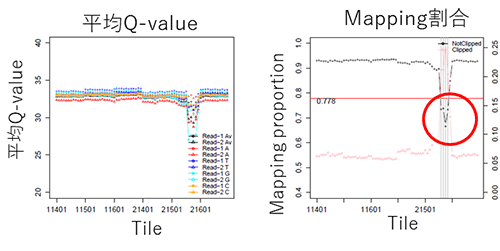
不良tileがある場合、csDAIでは不良tileデータを除いてBAMファイルを作成します。
変異解析
左右スクロールで表全体を閲覧できます
| 項目 | 内容 |
|---|---|
|
Germline call |
GATKのHaplotypeCaller + GenomicsDBImport + GenotypeGVCFsを使用*1 |
|
Somatic call |
GATKのMutect2を使用*2 |
|
Germline CNV |
GATKのGermlineCNVCaller*3を使用。 |
|
Somatic CNV |
GATKのDenoiseReadCounts + ModelSegments + CallCopyRatioSegmentsを使用*4 |
|
Structural variant |
Manta*5を使用 |
|
Fusion |
STAR-Fusion*6、Arriba*7、FusionCatcher*8を使用 |
|
Expression |
StringTieを使用*9 |
- *1)
- *2)
- *3)
- *4)
- *5)
- *6)
- *7)
- *8)
- *9)
アノテーション
csDAIではアノテーションを変異毎に付与しています。そのためVCFファイル中のmultiple alleleは複数行に分解されて出力されます。

スプレッドシート等で一覧表示するためのテキストファイル(タブ区切り)とPDFのアノテーションレポートが出力されます。アノテーション項目が多いため、スプレッドシート等で効率的なフィルタリングを行うために様々な集団中での最大頻度であるMaxAAFと変異を独自にカテゴリー化したdeleterious flagを付与しています。
Deleterious flag一覧
左右スクロールで表全体を閲覧できます
| Flag | 条件 |
|---|---|
|
E |
SnpEff*1のPutative_impact(csDAIではPutativeImpact)でHIGHと判定 |
|
D |
ClinVar*2においてPathogenic, Likely pathogenic, drug response、Conflicting_interpretations_of_pathogenicityで且つCLNSIGCONFに「PathogenicもしくはLikely pathogenic」がある、(OPTION)HGMD®*3のVariant_classにDM若しくはDM?が付与 |
|
S |
Exon中のmulti-nucleotide variant(MNV) |
|
H |
Exon中で遺伝子型callがphase情報付きでcallされたサンプルがある(0/1ではなく0|1の様なcall) |
|
C |
COSMIC*4においてConfirmed somatic variantと登録されている |
|
N |
InterProScan*5でドメイン予測された領域中のnon-frameshift insertion/deletion |
|
M |
InterProScan*5でドメイン予測された領域中のnonsynonymous変異 |
|
T |
SpliceAI *6(SNV, 1 base insertions and 1-4 base deletions only)のスコア0.2以上 |
|
A |
EnhancerAtlas*7もしくはHACER*8にヒットしたindel |
|
O |
Exonicの6塩基以上のindel、もしくはRepeatMasker*9でtRNA,snRNA,scRNA,srpRNA以外のrepeatと判定されたexonic以外の領域での10塩基以上のindel |
|
P |
dbNSFP*10(dbscSNV及びregsnpを含む)の予測において一つでもD判定 |
|
NA |
上記以外(ただしNAとなる変異を出力していない場合がある)。 |
- *1)
- *2)
- *3)
- *4)
- *5)
- *6)
- *7)
- *8)
- *9)
- *10)
アノテーションレポートの例

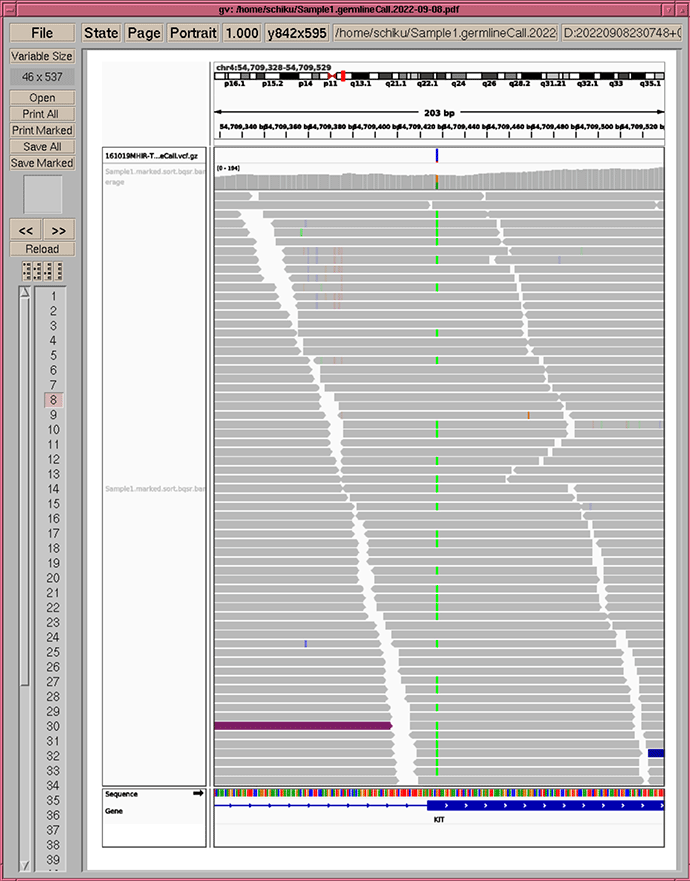
Utility tools
Utility toolsでは解析を補助する様々な機能を提供しています。Utility toolsタブを選択し、「Select tool」から使用するツールを選択します。
Utility toolsの選択画面
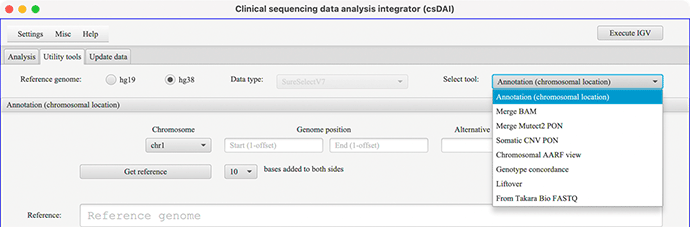
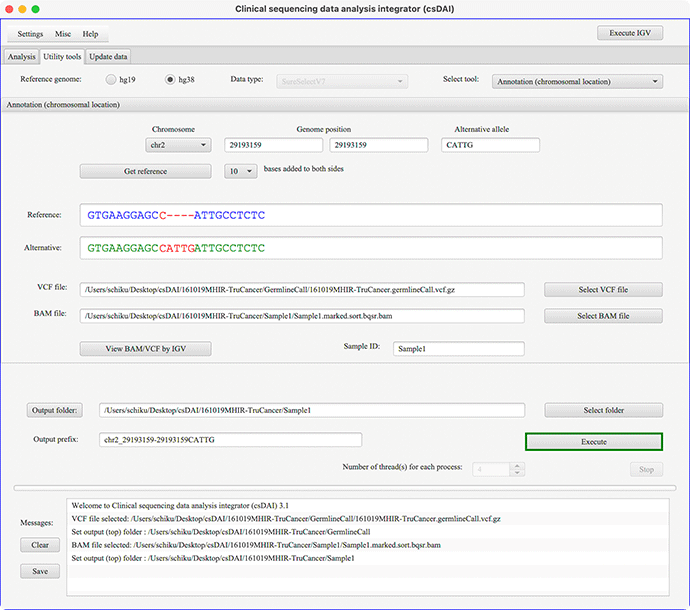
Annotation(chromosomal location)の画面例
Update data
定期的に更新される参照データやユーザーが設定するローカルデータについては、ユーザー様にデータ更新をお願いしています。
更新機能一覧
左右スクロールで表全体を閲覧できます
| データ | 概要 |
|---|---|
|
Bait |
新たなパネルを定義するbait領域ファイル(bedファイル)を登録します。 |
|
COSMIC*1 |
COSMICのデータを更新します。COSMICのライセンスはユーザー様にご用意頂きます。 |
|
ClinVar*2 |
ClinVarのデータを更新します。 |
|
HGMD®*3 |
HGMDデータの更新をします。HGMDのダウンロードライセンスが必要であり、QIAGEN社から購入する必要があります。 |
|
User reference allele frequencies |
アノテーションに付与するallele頻度データをINFOフィールドに含んだVCFファイルを指定します。 |
|
User genotype call counts |
アノテーションに付与するサンプル集団のVCFファイルを指定します。遺伝子型カウントと該当alleleにサンプルIDのリストが付きます。 |
COSMICデータの更新画面
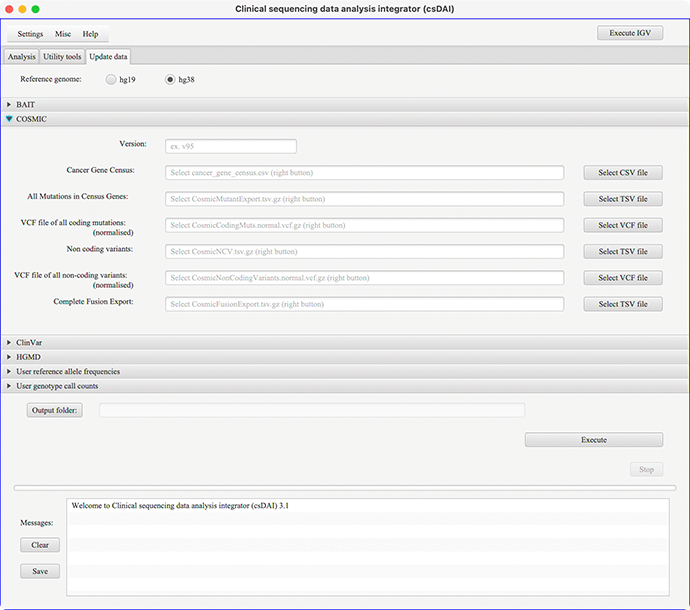
- *1)
- *2)
- *3)
- ※HGMDは米国QIAGENの米国およびその他の国における登録商標または商標です。