
Frequently Asked Questions
Q.1)何故IlluminaのシステムでFASTQファイルのアダプター配列を削除してはいけないのでしょうか?
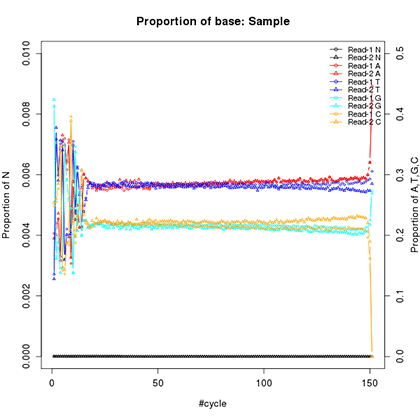
Illuminaのシステムではreadの末端において1塩基でもアダプター配列とマッチするとマッチした配列を除いてしまいます。測定するDNA断片のinsert長がNGSで読んでいるread長よりも短い場合はpaired-end readsではアダプター配列に相当するペアの両末端を除くとreverse complementary sequenceとなるため、readの末端がアダプター配列由来の配列なのか、シーケンシングしたサンプル由来なのか判断できるのですが、この処理を行っておりません。そのためquality control解析の塩基組成でreadの末端部分で大きな歪みが生じてしまいます。
Illminaのシステムでアダプター配列を除いた場合のcycle数毎の塩基組成の割合の例
Q.2)HaplotypeCallerによるgermline callで、reference homoとcallされているサンプルと変異がcallされているサンプルでread数の合計(depth)がかなり異なります。
csDAIではGVCF形式のg.vcfファイルをサンプルごとに作成し、GVCFから遺伝子型callを行っています。GVCF形式ではreferenceと比較して変異がない領域についてはブロックとして尤度情報を保持しています。この時depthについてはそのブロックの最低値を保持しており、遺伝子型をcallする際には保守的にこの最低値が付与されます。一方で変異があった箇所についてはきちんとdepth情報を保持しているため、変異があるサンプルのみ正しいdepthが付与されます。IGVでBAMファイルを閲覧するとreference homoとcallされたサンプルについても正しいdepth情報が得られますが、GVCFで保持されているdepthは様々な“Read Filters”を通過したread数のため、IGVで表示されるread数とは一般には一致しません。
Q.3)アノテーションCSVファイルから変異call結果をIGVで確認しようとBAMファイルを閲覧すると、readのマッピング状況とcall結果が異なっています。
BAMファイルはbwa memによるアライメントの結果であり、変異call結果はlocal de-novo assembly後に改めてreference genomeとアライメントした結果となります。Local de-novo assemblyによりread配列を結合して長い配列としてアライメントすることによりread長を超えるようなindelを検出することが可能になります。小さな領域でもGATKではindelを積極的に取り入れるアライメントパラメーターを使用している(多くの場合gap open penaltyの方がミスマッチよりも大きなペナルティとなるスコア体系が用いられる)と思われ、例えばTGAGAAGGとTGGAGGのアライメントでは下記のような違いが生じています。
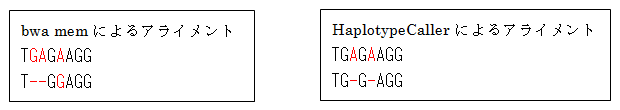
Q.4)変異call結果をIGVによりBAMファイルのアライメント結果から確認する場合、どのような疑わしい変異callが知られているのでしょうか?
エラー変異callの代表例としてはミスマッピング由来の変異callとシーケンシングエラー由来の変異callがあります。これらの変異callは、callされた変異は全てのサンプルでヘテロとしてcallされるのでHardy-Weinberg平衡からの乖離が観察されます。Segmental duplicationとアノテーションされている領域などでは特に注意が必要でしょう。またreadのストランドバイアスが生じていることも多いのですが、キャプチャーによるターゲットシーケンシングの場合にはターゲット領域の両端では片側からのreadしか取れていないこともあるので注意が必要です。
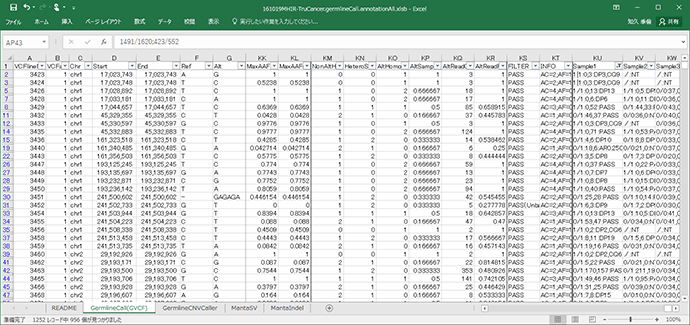
Q.5)Germline callでは複数サンプルによるcallを推奨している様ですが、ある特定のサンプルのみに興味がある場合はどの様に変異callを抽出すれば良いのでしょうか?
いくつかのやり方がありますが、Microsoft®社のExcel*®の「フィルター」機能を用いた例を示します。Excelでアノテーションテキストファイルを開いていただき、一番上の行を選択して「ホーム→並び替えとフィルター→フィルター」を選択してフィルター機能を有効にさせます。続いて注目しているサンプルの遺伝子型call結果(ファイルの右端の方にあります)があるサンプルIDのカラムから「テキストフィルター→指定の値を含まない」を選んで頂き、「NT」を含まない様に設定して頂ければ、そのサンプルに変異callがあり且つアノテーションの対象となった行だけが表示されます。
フィルターから「テキストフィルター→指定の値を含まない」を選択しているところ
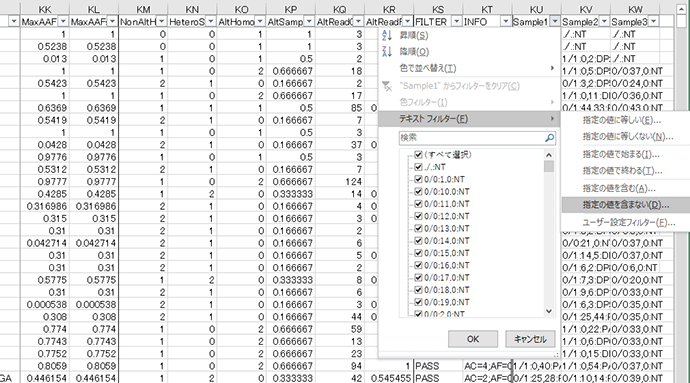
オートフィルタ―オプションの設定
フィルターを用いて「NT」を含まない結果を表示させた例
Q.6)HaplotypeCallerは複数サンプルで変異callを行うことにより変異callおよび遺伝子型callの精度を上げる仕組みがありますが、Mutect2はがん部と非がん部の複数のペアによる変異callは行えないのでしょうか?
GATK 4.1.0.0からmultiple samplesによるjoint callingがサポートされたとありますが、同じ患者由来の複数検体からのcallの様です。現在のcsDAIではこの機能を提供しておらず、がん部と非がん部のそれぞれ1つのBAMファイルからでしか変異callを行うことはできません。腫瘍内ヘテロ不均一性の解析を行いたい場合にはお手数ですが必要なT/Nペア毎に別々にcallしていただくことになります。
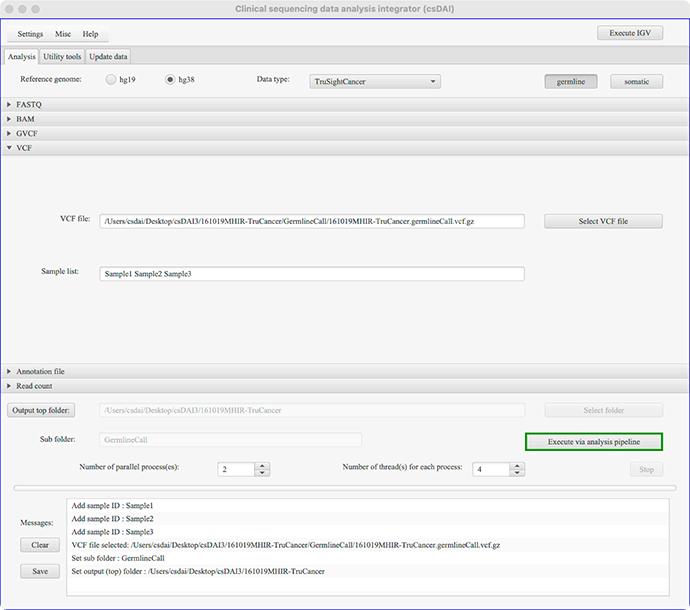
Q.7)VCFファイル中に含まれるサンプルIDを確認したい。
csDAIを起動し「Analysis」タブから「VCF」を開き「Select VCF file」から確認したいVCFファイルを選んでください。VCFファイル中のサンプルIDが「Sample list:」欄に表示されます。
csDAIを起動し「Analysis」タブから「VCF」を開き「Select VCF file」から確認したいVCFファイルを選んでサンプルIDを表示させたところ。本例ではSample1、Sample2、Sample3の3サンプルが含まれている。
Q.8)アノテーション情報にあるEnsemblとRefSeqは何が違うのでしょうか?
csDAIのアノテーション情報におけるEnsemblとはEuropean Bioinformatics Instituteから提供されているヒトゲノム上の遺伝子アノテーションのことになります。ヒト遺伝子に関してはEnsemblの遺伝子領域同定プログラムによる自動アノテーション結果とHavanaグループによるマニュアルキュレーションの結果を統合した結果になります(Ensemblはヒト以外の様々な生物種のゲノムも提供しています)。一方でcsDAIのアノテーション情報におけるRefSeqとは米国のNational Center for Biotechnology Information(NCBI)によるヒトゲノム上の遺伝子アノテーションのことを指しています。RefSeqGeneは「reference standards for well-characterized genes」の提供を目指しており、このmRNA配列をゲノムにマップした結果をヒトゲノム上の遺伝子アノテーションとして使用しています。このためEnsemblと比較してsplicing variantの数は少なく、またゲノム上の複数個所にマップされている遺伝子もあります。
Q.9)染色体上の位置の表現方法に1-based coordinate system(1-based)と0-based coordinate system(0-based)がある様ですが、どのような違いがあるのでしょうか。またどのフォーマットが1-basedであり、0-basedなのでしょうか?
1-based は塩基を1から順番に数えた形式であり、0-basedは下記において塩基と塩基の間の「|」を0から順番に数えた形式になります(UCSC Genome Browserの形式)。
左右スクロールで表全体を閲覧できます
|
1-based: |
|
1 |
|
2 |
|
3 |
|
4 |
|
5 |
|
6 |
|
|
|
| |
A |
| |
T |
| |
G |
| |
C |
| |
A |
| |
T |
| |
|
0-based: |
0 |
|
1 |
|
2 |
|
3 |
|
4 |
|
5 |
|
6 |
0-basedの場合、配列は間の「|」の番号で表現することになり、1塩基でもstartとendを指定します。例えば上記で1-basedで3番目のG塩基は、0-basedではstart=2、end=3となります。一般に0-basedと1-basedでは開始位置は1だけずれますが、終了位置は同じになります。計算機による表現では0-basedの方がメリットは多く、例えばinsertionを表現する場合1-basedでは「3のGをGAに置換」のような表現になりますが、0-basedでは「3の位置にAを挿入」と表現できます。また配列長を計算する場合も0-basedでは「#end-#start」で計算できますが、1-basedでは「#end-#start+1」となります。
csDAIで使われているファイルフォーマットでは、SAM、VCF、annotation.txt、アノテーションファイルは1-basedですが、BED、BAMは0-basedとなっています。
Q.10)Phredスコアとは何でしょうか?
何らかの方法で計算したp値(区間[0,1])を-10 log10pと変換した値になります。元々Phred quality scoreとはシーケンシングデータ解析プログラムPhredが出力した塩基のcallの精度を表していました。FASTQ形式のquality valueはcallされた塩基が間違いである(推定)確率pのPhredスコアに33を足したiASCIIコード(実際には2~41に対応する#$%&'()*+,-./0123456789:;=<=>?@ABCDEFGHIの文字列)で表現されています。
- ※MicrosoftおよびExcelは、米国Microsoft Corporationの米国およびその他の国における登録商標です。